Welcome to scCODA’s documentation!¶
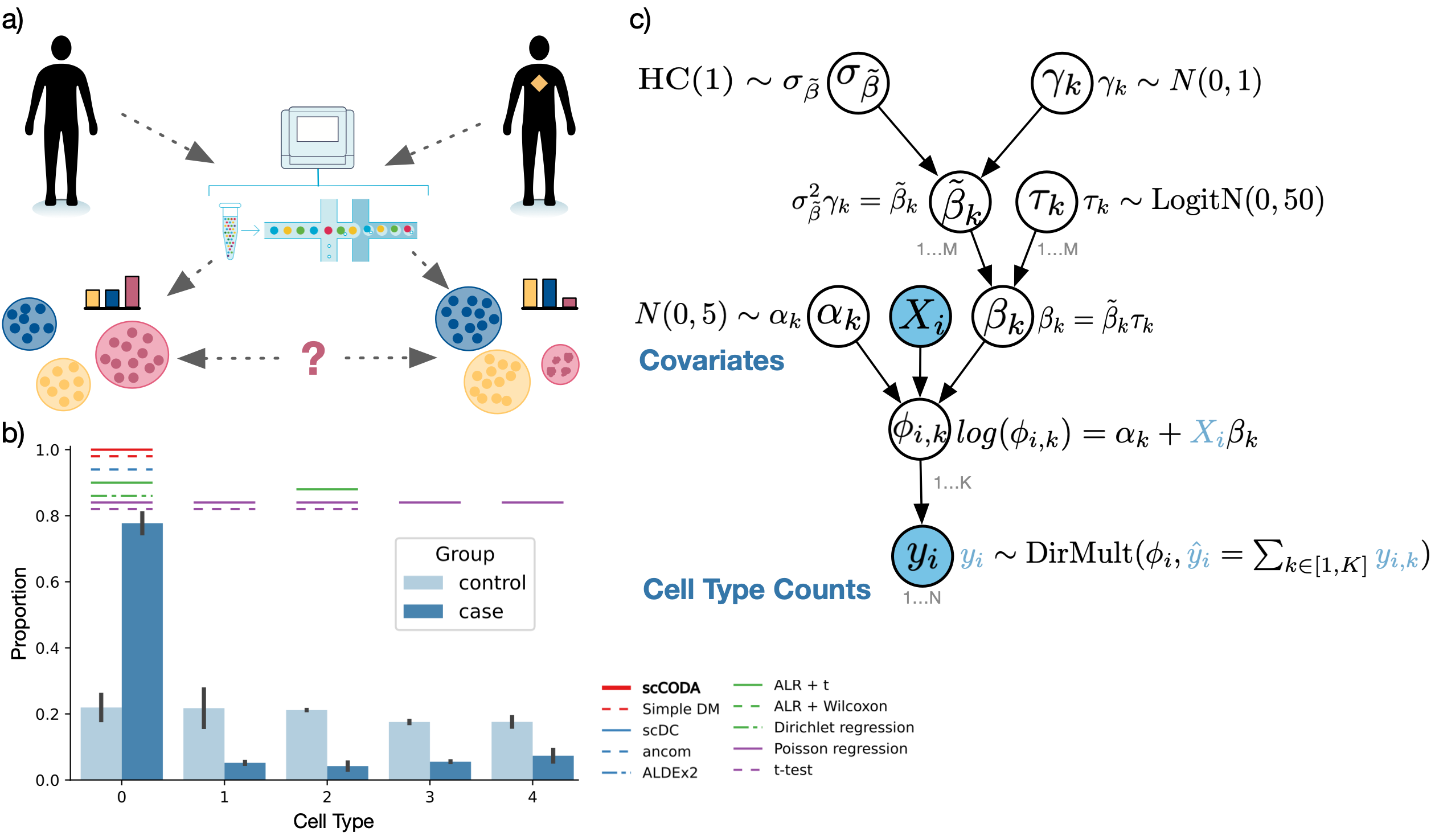
scCODA is a toolbox for statistical models to analyze changes in compositional data, especially from single-cell RNA-seq experiments. Its main purpose is to provide a platform and implementation for the scCODA model, which is described by Büttner, Ostner et al..
The package is available on github.
Please also check out the tutorials that explain the most important functionalities.
Motivation¶
When analyzing biological processes via single-cell RNA-sequencing experiments, it is often of interest to assess how cell populations change under one or more conditions. This task, however, is non-trivial, as there are several limitations that have to be addressed:
scRNA-seq population data is compositional. This must be considered to avoid an inflation of false-positive results.
Most datasets consist only of very few samples, making frequentist tests inaccurate.
A condition usually only effects a fraction of cell types. Therefore, sparse effects are preferable.
The scCODA model overcomes all these limitations in a fully Bayesian model, that outperforms other compositional and non-compositional methods.
scCODA is fully integrable with scanpy, but provides its own data structure for aggregating, plotting and analyzing compositional data from scRNA-seq. Additionally to the scCODA model, the package also features a variety of implementations of other statistical models that can be used as comparisons.
Contents
Reference¶
Büttner, Ostner et al. (2021), scCODA is a Bayesian model for compositional single-cell data analysis NatComms.