Modeling options and result analysis in scCODA¶
This tutorial notebook serves as an extension to the general tutorial and presents ways to alternate the model and perform more in-depth result analysis and diagnostics. We will focus on:
Modifications of the model formula and reference cell type to perform different modeling tasks
Inference methods available in scCODA
Advanced interpretation and analysis of results
Alternative differential abundance testing using all references
We will again analyze the small intestinal epithelium data of mice from Haber et al., 2017. First, we read in the data and perform the same preprocessing steps as in the general tutorial:
[1]:
# Setup
import warnings
warnings.filterwarnings("ignore")
import pandas as pd
import matplotlib.pyplot as plt
import arviz as az
from sccoda.util import comp_ana as mod
from sccoda.util import cell_composition_data as dat
from sccoda.util import data_visualization as viz
[2]:
# Load data
cell_counts = pd.read_csv("../data/haber_counts.csv")
# Convert data to anndata object
data_all = dat.from_pandas(cell_counts, covariate_columns=["Mouse"])
# Extract condition from mouse name and add it as an extra column to the covariates
data_all.obs["Condition"] = data_all.obs["Mouse"].str.replace(r"_[0-9]", "")
print(f"Entire dataset: {data_all}")
# Select control and salmonella data
data_salm = data_all[data_all.obs["Condition"].isin(["Control", "Salm"])].copy()
print(f"Salmonella dataset: {data_salm}")
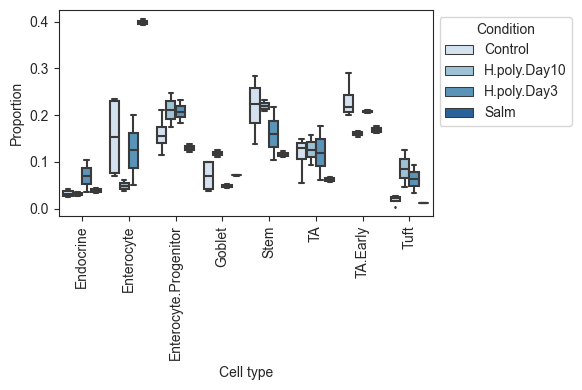
viz.boxplots(data_all, feature_name="Condition")
plt.show()
Entire dataset: AnnData object with n_obs × n_vars = 10 × 8
obs: 'Mouse', 'Condition'
Salmonella dataset: AnnData object with n_obs × n_vars = 6 × 8
obs: 'Mouse', 'Condition'
Tweaking the model formula and reference cell type¶
First, we take a closer look at how changing the formula parameter of the scCODA model influences the results. Internally, the formula string is converted into a linear model-like design matrix via patsy, which has a similar syntax to the lm function in the R language.
Multi-level categories¶
Patsy allows us to automatically handle categorical covariates, even with multiple levels. For example, we can model the effect of all three diseases at once:
[3]:
# model all three diseases at once
model_all = mod.CompositionalAnalysis(data_all, formula="Condition", reference_cell_type="Endocrine")
all_results = model_all.sample_hmc()
all_results.summary()
2021-11-28 19:15:21.670317: I tensorflow/core/platform/cpu_feature_guard.cc:142] This TensorFlow binary is optimized with oneAPI Deep Neural Network Library (oneDNN) to use the following CPU instructions in performance-critical operations: AVX2 FMA
To enable them in other operations, rebuild TensorFlow with the appropriate compiler flags.
WARNING:tensorflow:From /Users/johannes.ostner/opt/anaconda3/envs/scCODA_3/lib/python3.8/site-packages/tensorflow/python/ops/array_ops.py:5043: calling gather (from tensorflow.python.ops.array_ops) with validate_indices is deprecated and will be removed in a future version.
Instructions for updating:
The `validate_indices` argument has no effect. Indices are always validated on CPU and never validated on GPU.
2021-11-28 19:15:23.943628: I tensorflow/compiler/mlir/mlir_graph_optimization_pass.cc:176] None of the MLIR Optimization Passes are enabled (registered 2)
0%| | 0/20000 [00:00<?, ?it/s]2021-11-28 19:15:25.649198: I tensorflow/compiler/xla/service/service.cc:169] XLA service 0x7fd21d1c63a0 initialized for platform Host (this does not guarantee that XLA will be used). Devices:
2021-11-28 19:15:25.649348: I tensorflow/compiler/xla/service/service.cc:177] StreamExecutor device (0): Host, Default Version
2021-11-28 19:15:28.022894: I tensorflow/compiler/jit/xla_compilation_cache.cc:337] Compiled cluster using XLA! This line is logged at most once for the lifetime of the process.
100%|██████████| 20000/20000 [01:38<00:00, 203.36it/s]
MCMC sampling finished. (123.099 sec)
Acceptance rate: 78.2%
Compositional Analysis summary:
Data: 10 samples, 8 cell types
Reference index: 0
Formula: Condition
Intercepts:
Final Parameter Expected Sample
Cell Type
Endocrine 0.986 46.634633
Enterocyte 1.905 116.902874
Enterocyte.Progenitor 2.348 182.061302
Goblet 1.474 75.970374
Stem 2.427 197.027528
TA 1.878 113.788726
TA.Early 2.534 219.278684
Tuft 0.626 32.535879
Effects:
Final Parameter \
Covariate Cell Type
Condition[T.H.poly.Day10] Endocrine 0.000000
Enterocyte 0.000000
Enterocyte.Progenitor 0.000000
Goblet 0.000000
Stem 0.000000
TA 0.000000
TA.Early 0.000000
Tuft 0.000000
Condition[T.H.poly.Day3] Endocrine 0.000000
Enterocyte 0.000000
Enterocyte.Progenitor 0.000000
Goblet 0.000000
Stem 0.000000
TA 0.000000
TA.Early 0.000000
Tuft 0.000000
Condition[T.Salm] Endocrine 0.000000
Enterocyte 1.554932
Enterocyte.Progenitor 0.000000
Goblet 0.000000
Stem 0.000000
TA 0.000000
TA.Early 0.000000
Tuft 0.000000
Expected Sample \
Covariate Cell Type
Condition[T.H.poly.Day10] Endocrine 46.634633
Enterocyte 116.902874
Enterocyte.Progenitor 182.061302
Goblet 75.970374
Stem 197.027528
TA 113.788726
TA.Early 219.278684
Tuft 32.535879
Condition[T.H.poly.Day3] Endocrine 46.634633
Enterocyte 116.902874
Enterocyte.Progenitor 182.061302
Goblet 75.970374
Stem 197.027528
TA 113.788726
TA.Early 219.278684
Tuft 32.535879
Condition[T.Salm] Endocrine 32.304092
Enterocyte 383.418044
Enterocyte.Progenitor 126.114963
Goblet 52.625137
Stem 136.482158
TA 78.822138
TA.Early 151.895669
Tuft 22.537800
log2-fold change
Covariate Cell Type
Condition[T.H.poly.Day10] Endocrine 0.000000
Enterocyte 0.000000
Enterocyte.Progenitor 0.000000
Goblet 0.000000
Stem 0.000000
TA 0.000000
TA.Early 0.000000
Tuft 0.000000
Condition[T.H.poly.Day3] Endocrine 0.000000
Enterocyte 0.000000
Enterocyte.Progenitor 0.000000
Goblet 0.000000
Stem 0.000000
TA 0.000000
TA.Early 0.000000
Tuft 0.000000
Condition[T.Salm] Endocrine -0.529685
Enterocyte 1.713608
Enterocyte.Progenitor -0.529685
Goblet -0.529685
Stem -0.529685
TA -0.529685
TA.Early -0.529685
Tuft -0.529685
Different reference levels¶
Per default, categorical variables are encoded via full-rank treatment coding. Hereby, the value of the first sample in the dataset is used as the default (control) category. We can select the default level by changing the model formula to "C(<CovariateName>, Treatment('<ReferenceLevelName>'))":
For example, we can switch the salmonella model to test diseased versus healthy samples, which switches the sign of the only credible effect (Enterocytes).
[4]:
# Set salmonella infection as "default" category
model_salm_switch_cond = mod.CompositionalAnalysis(data_salm, formula="C(Condition, Treatment('Salm'))", reference_cell_type="Goblet")
switch_results = model_salm_switch_cond.sample_hmc()
switch_results.summary()
100%|██████████| 20000/20000 [01:02<00:00, 317.86it/s]
MCMC sampling finished. (82.254 sec)
Acceptance rate: 49.8%
Compositional Analysis summary:
Data: 6 samples, 8 cell types
Reference index: 3
Formula: C(Condition, Treatment('Salm'))
Intercepts:
Final Parameter Expected Sample
Cell Type
Endocrine 1.269 28.559849
Enterocyte 3.708 327.340808
Enterocyte.Progenitor 2.566 104.480647
Goblet 1.777 47.465439
Stem 2.615 109.727702
TA 2.048 62.240247
TA.Early 2.877 142.594061
Tuft 0.450 12.591247
Effects:
Final Parameter \
Covariate Cell Type
C(Condition, Treatment('Salm'))[T.Control] Endocrine 0.000000
Enterocyte -1.365487
Enterocyte.Progenitor 0.000000
Goblet 0.000000
Stem 0.000000
TA 0.000000
TA.Early 0.000000
Tuft 0.000000
Expected Sample \
Covariate Cell Type
C(Condition, Treatment('Salm'))[T.Control] Endocrine 40.336384
Enterocyte 118.009650
Enterocyte.Progenitor 147.562807
Goblet 67.037616
Stem 154.973463
TA 87.904755
TA.Early 201.392129
Tuft 17.783195
log2-fold change
Covariate Cell Type
C(Condition, Treatment('Salm'))[T.Control] Endocrine 0.498093
Enterocyte -1.471889
Enterocyte.Progenitor 0.498093
Goblet 0.498093
Stem 0.498093
TA 0.498093
TA.Early 0.498093
Tuft 0.498093
Switching the reference cell type¶
Compositional analysis generally does not allow statements on absolute abundance changes, but only in relation to a reference category, which is assumed to be unchanged in absolute abundance. The reference cell type fixes this category in scCODA. Thus, an interpretation of scCODA’s effects should always be formulated like:
“Using cell type xy as a reference, cell types (a, b, c) were found to credibly change in abundance”
Switching the reference cell type might thus produce different results. For example, if we choose a different cell type as the reference (such as Enterocytes in the salmonella infection data), scCODA can find other credible effects on the other cell types.
[13]:
model_salm_ref = mod.CompositionalAnalysis(data_salm, formula="Condition", reference_cell_type="Enterocyte")
reference_results = model_salm_ref.sample_hmc()
reference_results.summary()
100%|██████████| 20000/20000 [01:02<00:00, 321.43it/s]
MCMC sampling finished. (80.035 sec)
Acceptance rate: 54.2%
Compositional Analysis summary:
Data: 6 samples, 8 cell types
Reference index: 1
Formula: Condition
Intercepts:
Final Parameter Expected Sample
Cell Type
Endocrine 0.563 34.927176
Enterocyte 2.088 160.495387
Enterocyte.Progenitor 1.870 129.058424
Goblet 1.100 59.755737
Stem 2.095 161.622796
TA 1.475 86.944084
TA.Early 2.220 183.142621
Tuft -0.043 19.053774
Effects:
Final Parameter Expected Sample \
Covariate Cell Type
Condition[T.Salm] Endocrine 0.0 34.927176
Enterocyte 0.0 160.495387
Enterocyte.Progenitor 0.0 129.058424
Goblet 0.0 59.755737
Stem 0.0 161.622796
TA 0.0 86.944084
TA.Early 0.0 183.142621
Tuft 0.0 19.053774
log2-fold change
Covariate Cell Type
Condition[T.Salm] Endocrine 0.0
Enterocyte 0.0
Enterocyte.Progenitor 0.0
Goblet 0.0
Stem 0.0
TA 0.0
TA.Early 0.0
Tuft 0.0
Inference algorithms in scCODA¶
Currently, scCODA performs parameter inference via Markov-chain Monte Carlo (MCMC) methods. There are three different MCMC sampling methods available for scCODA:
Hamiltonian Monte Carlo (HMC) sampling:
sample_hmc()HMC sampling with Dual-averaging step size adaptation (Nesterov, 2009):
sample_hmc_da()No-U-Turn sampling (Hoffman and Gelman, 2014):
sample_nuts()
Generally, it is recommended to use the standard HMC sampling. Other methods, such as variational inference, are in consideration.
For all MCMC sampling methods, properties such as the MCMC chain length and the number of burn-in samples are directly adjustable.
Result analysis and diagnostics¶
The “getting started” tutorial explains how to do interpret the basic output of scCODA. To follow this up, we now take a look at how MCMC diagnostics and more advanced result analysis in scCODA can be performed.
For this section, we again use the model of salmonella infection versus control group, with a reference cell type of Goblet cells.
[6]:
model_salm = mod.CompositionalAnalysis(data_salm, formula="Condition", reference_cell_type="Goblet")
salm_results = model_salm.sample_hmc(num_results=20000)
100%|██████████| 20000/20000 [01:03<00:00, 314.08it/s]
MCMC sampling finished. (80.894 sec)
Acceptance rate: 55.8%
Extended model summary¶
result.summary_extended() gives us, apart from the properties already explained in the basic tutorial, more information about the posterior inferred by the model. The extended summary also includes some information on the MCMC sampling procedure (chain length, burn-in, acceptance rate, duration).
For both effects and intercepts, we also get the standard deviation (SD) and high density interval endpoints of the posterior density of the generated Markov chain.
The effects summary also includes the spike-and-slab inclusion probability for each effect, i.e. the share of MCMC samples, for which this effect was not set to 0 by the spike-and-slab prior. A threshold on this value serves as the deciding factor whether an effect is considered statistically credible
We can also use the summary tables from summary_extended() as pandas DataFrames to tweak them further. They are also accessible as result.intercept_df and result.effect_df, respectively. Furthermore, the tables a direct result of the summary() function in arviz and support all its functionality. This means that we can, for example, change the credible interval:
[7]:
salm_results.summary_extended(hdi_prob=0.9)
Compositional Analysis summary (extended):
Data: 6 samples, 8 cell types
Reference index: 3
Formula: Condition
Spike-and-slab threshold: 1.000
MCMC Sampling: Sampled 20000 chain states (5000 burnin samples) in 80.894 sec. Acceptance rate: 55.8%
Intercepts:
Final Parameter HDI 5% HDI 95% SD \
Cell Type
Endocrine 1.105 0.548 1.750 0.369
Enterocyte 2.330 1.842 2.844 0.312
Enterocyte.Progenitor 2.517 2.055 3.071 0.310
Goblet 1.749 1.253 2.277 0.316
Stem 2.710 2.229 3.235 0.306
TA 2.114 1.576 2.658 0.330
TA.Early 2.858 2.361 3.341 0.297
Tuft 0.433 -0.210 1.065 0.394
Expected Sample
Cell Type
Endocrine 34.199308
Enterocyte 116.420125
Enterocyte.Progenitor 140.359279
Goblet 65.118287
Stem 170.239355
TA 93.803805
TA.Early 197.394727
Tuft 17.465114
Effects:
Final Parameter HDI 5% HDI 95% \
Covariate Cell Type
Condition[T.Salm] Endocrine 0.000000 -0.432 1.033
Enterocyte 1.347912 0.912 1.768
Enterocyte.Progenitor 0.000000 -0.255 0.581
Goblet 0.000000 0.000 0.000
Stem 0.000000 -0.720 0.129
TA 0.000000 -0.789 0.249
TA.Early 0.000000 -0.331 0.422
Tuft 0.000000 -0.772 0.866
SD Inclusion probability \
Covariate Cell Type
Condition[T.Salm] Endocrine 0.348 0.506933
Enterocyte 0.265 1.000000
Enterocyte.Progenitor 0.158 0.334800
Goblet 0.000 0.000000
Stem 0.214 0.398667
TA 0.247 0.418933
TA.Early 0.119 0.267467
Tuft 0.325 0.417733
Expected Sample log2-fold change
Covariate Cell Type
Condition[T.Salm] Endocrine 24.475708 -0.482617
Enterocyte 320.727867 1.462009
Enterocyte.Progenitor 100.452112 -0.482617
Goblet 46.603755 -0.482617
Stem 121.836638 -0.482617
TA 67.133362 -0.482617
TA.Early 141.271153 -0.482617
Tuft 12.499406 -0.482617
Diagnostics and plotting¶
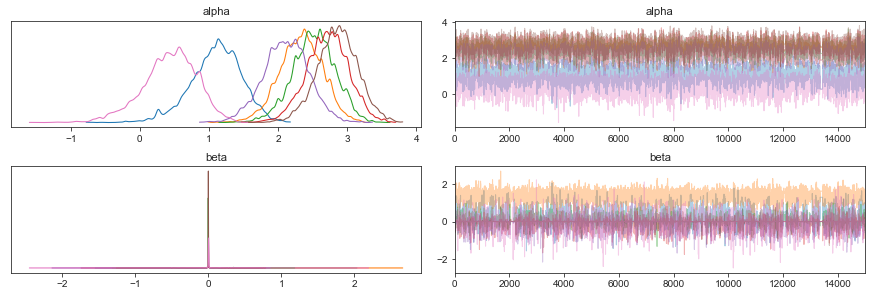
Similarly to the summary dataframes being compatible with arviz, the result class itself is an extension of arviz’s Inference Data class. This means that we can use all its MCMC diagnostic and plotting functionality. As an example, looking at the MCMC trace plots and kernel density estimates, we see that they are indicative of a well sampled MCMC chain:
Note: Due to the spike-and-slab priors, the beta parameters have many values at 0, which looks like a convergence issue, but is actually not.
Caution: Trying to plot a kernel density estimate for an effect on the reference cell type results in an error, since it is constant at 0 for the entire chain. To avoid this, add coords={"cell_type": salm_results.posterior.coords["cell_type_nb"]} as an argument to ``az.plot_trace``, which causes the plots for the reference cell type to be skipped.
[8]:
az.plot_trace(
salm_results,
divergences=False,
var_names=["alpha", "beta"],
coords={"cell_type": salm_results.posterior.coords["cell_type_nb"]},
)
plt.show()
Using all cell types as reference alternatively to reference selection¶
scCODA uses a reference cell type that is considered to be unchanged over the experiment to guarantee the unique identifiability of results. If no such cell type is known beforehand, setting reference_cell_type="automatic" will find a suited reference. Alternatively, it is possible to find credible effects on cell types that are mostly independent of the reference. By sequentially running scCODA and selecting each cell type as the reference once, we can then use a majority vote to find the
cell types that were credibly changing more than half of the time.
Below, an example code for this procedure on the Salmonella infection data shows that only Enterocytes were found to be credible more than half of the time. Indeed, they re credibly changing for every reference cell type except themselves. All other cell types were not found to change with any reference.
[9]:
# Run scCODA with each cell type as the reference
cell_types = data_salm.var.index
results_cycle = pd.DataFrame(index=cell_types, columns=["times_credible"]).fillna(0)
for ct in cell_types:
print(f"Reference: {ct}")
# Run inference
model_temp = mod.CompositionalAnalysis(data_salm, formula="Condition", reference_cell_type=ct)
temp_results = model_temp.sample_hmc(num_results=20000)
# Select credible effects
cred_eff = temp_results.credible_effects()
cred_eff.index = cred_eff.index.droplevel(level=0)
# add up credible effects
results_cycle["times_credible"] += cred_eff.astype("int")
Reference: Endocrine
100%|██████████| 20000/20000 [01:03<00:00, 313.42it/s]
WARNING:tensorflow:5 out of the last 5 calls to <function CompositionalModel.sampling.<locals>.sample_mcmc at 0x7fd1ed75e3a0> triggered tf.function retracing. Tracing is expensive and the excessive number of tracings could be due to (1) creating @tf.function repeatedly in a loop, (2) passing tensors with different shapes, (3) passing Python objects instead of tensors. For (1), please define your @tf.function outside of the loop. For (2), @tf.function has experimental_relax_shapes=True option that relaxes argument shapes that can avoid unnecessary retracing. For (3), please refer to https://www.tensorflow.org/guide/function#controlling_retracing and https://www.tensorflow.org/api_docs/python/tf/function for more details.
MCMC sampling finished. (81.026 sec)
Acceptance rate: 50.1%
Reference: Enterocyte
100%|██████████| 20000/20000 [01:04<00:00, 309.59it/s]
WARNING:tensorflow:6 out of the last 6 calls to <function CompositionalModel.sampling.<locals>.sample_mcmc at 0x7fd1ed75e3a0> triggered tf.function retracing. Tracing is expensive and the excessive number of tracings could be due to (1) creating @tf.function repeatedly in a loop, (2) passing tensors with different shapes, (3) passing Python objects instead of tensors. For (1), please define your @tf.function outside of the loop. For (2), @tf.function has experimental_relax_shapes=True option that relaxes argument shapes that can avoid unnecessary retracing. For (3), please refer to https://www.tensorflow.org/guide/function#controlling_retracing and https://www.tensorflow.org/api_docs/python/tf/function for more details.
MCMC sampling finished. (85.399 sec)
Acceptance rate: 56.5%
Reference: Enterocyte.Progenitor
100%|██████████| 20000/20000 [01:08<00:00, 291.32it/s]
MCMC sampling finished. (93.835 sec)
Acceptance rate: 60.7%
Reference: Goblet
100%|██████████| 20000/20000 [01:13<00:00, 271.64it/s]
MCMC sampling finished. (90.710 sec)
Acceptance rate: 58.2%
Reference: Stem
100%|██████████| 20000/20000 [01:07<00:00, 295.02it/s]
MCMC sampling finished. (86.994 sec)
Acceptance rate: 51.8%
Reference: TA
100%|██████████| 20000/20000 [01:22<00:00, 242.56it/s]
MCMC sampling finished. (103.528 sec)
Acceptance rate: 53.4%
Reference: TA.Early
100%|██████████| 20000/20000 [01:21<00:00, 244.23it/s]
MCMC sampling finished. (105.065 sec)
Acceptance rate: 54.0%
Reference: Tuft
100%|██████████| 20000/20000 [01:23<00:00, 240.64it/s]
MCMC sampling finished. (104.704 sec)
Acceptance rate: 49.7%
[10]:
# Calculate percentages
results_cycle["pct_credible"] = results_cycle["times_credible"]/len(cell_types)
results_cycle["is_credible"] = results_cycle["pct_credible"] > 0.5
print(results_cycle)
times_credible pct_credible is_credible
Endocrine 0 0.000 False
Enterocyte 7 0.875 True
Enterocyte.Progenitor 0 0.000 False
Goblet 0 0.000 False
Stem 0 0.000 False
TA 0 0.000 False
TA.Early 0 0.000 False
Tuft 0 0.000 False
[10]: