Using other compositional differentiation methods in scCODA¶
While the scCODA package is mainly aimed at providing an implementation and interface of the scCODA model (Büttner, Ostner et al., 2021), the module sccoda.models.other_models contains a variety of wrappers for other compositional differentiation methods that can be used with scCODA’s data handling pipeline.
The available methods are:
A fully Bayesian Dirichlet-Multinomial model without spike-and-slab priors.
The SCDC model (Cao et al., 2019)
A (non-compositional) Poisson regression model
A model using the centered logratio (CLR) transform together with a generalized linear model
A (non-compositional) t-test
A CLR transform together with a t-test
The ALDEx2 model for compositional differentiation in microbial data (Fernandes et al., 2014)
An additive logratio (ALR) transform of the data paired with a t-test
An ALR transform of the data paired with a Wilcoxon rank-sum test
The ANCOM model for compositional differentiation in microbial data (Mandal et al., 2015)
The Bias-corrected ANCOM model for compositional differentiation in microbial data (Lin, Peddada, 2020]
A Dirichlet regression approach (Maier, 2014)
Beta-Binomial modeling via corncob (Martin e al., 2019)
Note: Some methods (SCDC, ALDEx2, Dirichlet Regression, ANCOMBC, Beta-Binomial) require an R environment with the according packages installed
NOTE: If you want to run this tutorial, make sure to have `scikit-bio <https://github.com/biocore/scikit-bio>`__ installed. It is not automatically included when installing via pip.
This tutorial aims at showcasing the use of some of these methods.
Setup and data handling¶
We use the salmonella infection data from Haber et al., 2017.
[1]:
# Setup
import warnings
warnings.filterwarnings("ignore")
import pandas as pd
import patsy as pt
import numpy as np
from sccoda.model import other_models as om
from sccoda.util import cell_composition_data as dat
from sccoda.util import data_visualization as viz
[2]:
# Get some data (Salmonella infection from Haber et al.)
cell_counts = pd.read_csv("../data/haber_counts.csv")
# Convert data to anndata object
data_all = dat.from_pandas(cell_counts, covariate_columns=["Mouse"])
# Extract condition from mouse name and add it as an extra column to the covariates
data_all.obs["Condition"] = data_all.obs["Mouse"].str.replace(r"_[0-9]", "")
# Select control and salmonella data
data_salm = data_all[data_all.obs["Condition"].isin(["Control", "Salm"])]
print(data_salm)
View of AnnData object with n_obs × n_vars = 6 × 8
obs: 'Mouse', 'Condition'
[3]:
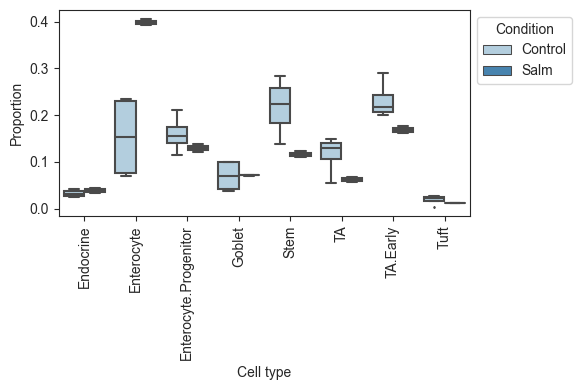
viz.boxplots(data_salm, feature_name="Condition")
[3]:
<AxesSubplot:xlabel='Cell type', ylabel='Proportion'>
The ancom model¶
Using the ancom model (Mandal et al., 2015) with scCODA is straightforward. We use the implementation in scikit-bio and also its output format:
The column “Reject null hypothesis” shows the columns that change significantly according to ancom
[4]:
ancom_model = om.AncomModel(data_salm, covariate_column="Condition")
ancom_model.fit_model()
print(ancom_model.ancom_out)
Trying to set attribute `.X` of view, copying.
( W Reject null hypothesis
Endocrine 0 False
Enterocyte 0 False
Enterocyte.Progenitor 0 False
Goblet 0 False
Stem 0 False
TA 0 False
TA.Early 0 False
Tuft 0 False, Percentile 0.0 25.0 50.0 75.0 100.0 0.0 25.0 \
Group Control Control Control Control Control Salm Salm
Endocrine 5.0 20.75 31.0 38.25 45.0 32.0 33.25
Enterocyte 46.0 55.75 78.5 128.75 221.0 332.0 342.25
Enterocyte.Progenitor 23.0 107.75 162.0 190.50 198.0 113.0 113.75
Goblet 20.0 32.00 36.0 58.00 124.0 59.0 61.00
Stem 50.0 110.75 185.0 241.75 250.0 90.0 96.75
TA 11.0 96.50 127.5 136.25 155.0 47.0 51.50
TA.Early 40.0 153.25 193.5 238.25 365.0 132.0 141.00
Tuft 4.0 4.75 11.5 21.75 33.0 10.0 10.50
Percentile 50.0 75.0 100.0
Group Salm Salm Salm
Endocrine 34.5 35.75 37.0
Enterocyte 352.5 362.75 373.0
Enterocyte.Progenitor 114.5 115.25 116.0
Goblet 63.0 65.00 67.0
Stem 103.5 110.25 117.0
TA 56.0 60.50 65.0
TA.Early 150.0 159.00 168.0
Tuft 11.0 11.50 12.0 )
The ALDEx2 model¶
The ALDEx2 model (Fernandes et al., 2014) requires an R installation with the ALDEx2 package available. The paths r_home and r_path must point to the installation folder and the R executable of your R version.
The result columns show the p-values of various tests (see the documentation of ALDEx2 for details)
[5]:
r_home = "/Library/Frameworks/R.framework/Resources"
r_path = "/Library/Frameworks/R.framework/Resources/bin/R"
[6]:
aldex2_model = om.ALDEx2Model(data_salm, covariate_column="Condition")
aldex2_model.fit_model(r_home=r_home, r_path=r_path)
print(aldex2_model.result)
R[write to console]: operating in serial mode
R[write to console]: computing center with all features
we.ep we.eBH wi.ep wi.eBH
1 0.385566 0.520241 0.519792 0.670258
2 0.055019 0.190518 0.133333 0.419444
3 0.554758 0.649357 0.742708 0.839306
4 0.549451 0.649280 0.890104 0.910625
5 0.045055 0.170609 0.146875 0.427778
6 0.211257 0.380195 0.436458 0.651677
7 0.165477 0.311701 0.277604 0.509593
8 0.644732 0.715048 0.594792 0.750084
The SCDC model¶
For using the SCDC model (Cao et al., 2019), an R environment with the package scdney (available on github) is required.
Using the model is similar to the other ones, the “p.values” column shows significances:
[7]:
scdc_model = om.scdney_model(data_salm, covariate_column="Condition")
result, _ = scdc_model.analyze(r_home=r_home, r_path=r_path)
print(result)
R[write to console]: ── Attaching packages ────────────────────────────────────────── scdney 0.2.0 ──
R[write to console]: ✔ scMerge 1.10.0 ✔ scClust 0.3.0
✔ scClassify 1.6.0 ✔ Cepo 1.0.0
✔ CiteFuse 1.6.0 ✔ scHOT 1.6.0
✔ scDC 0.1.0
R[write to console]: ── Attaching packages ─────────────────────────────────────── tidyverse 1.3.1 ──
R[write to console]: ✔ ggplot2 3.3.5 ✔ purrr 0.3.4
✔ tibble 3.1.6 ✔ dplyr 1.0.7
✔ tidyr 1.1.4 ✔ stringr 1.4.0
✔ readr 2.1.0 ✔ forcats 0.5.1
R[write to console]: ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ stringr::boundary() masks graph::boundary()
✖ dplyr::collapse() masks IRanges::collapse()
✖ dplyr::combine() masks Biobase::combine(), BiocGenerics::combine()
✖ dplyr::desc() masks IRanges::desc()
✖ tidyr::expand() masks S4Vectors::expand()
✖ dplyr::filter() masks stats::filter()
✖ dplyr::first() masks S4Vectors::first()
✖ dplyr::lag() masks stats::lag()
✖ ggplot2::Position() masks BiocGenerics::Position(), base::Position()
✖ purrr::reduce() masks IRanges::reduce()
✖ dplyr::rename() masks S4Vectors::rename()
✖ dplyr::select() masks AnnotationDbi::select(), MASS::select()
✖ dplyr::slice() masks IRanges::slice()
[1] "Calculating sample proportion..."
[1] "Calculating bootstrap proportion..."
[1] "Calculating BCa ..."
[1] "Calculating z0 ..."
[1] "Calculating acc ..."
[1] "fitting GLM... 10"
[1] "fitting GLM... 20"
[1] "fitting GLM... 30"
[1] "fitting GLM... 40"
[1] "fitting GLM... 50"
[1] "fitting GLM... 60"
[1] "fitting GLM... 70"
[1] "fitting GLM... 80"
[1] "fitting GLM... 90"
[1] "fitting GLM... 100"
term estimate std.error \
0 (Intercept) 3.340425 0.130826
1 cellTypescell_Enterocyte 1.318058 0.151683
2 cellTypescell_Enterocyte.Progenitor 1.573899 0.145437
3 cellTypescell_Goblet 0.643168 0.167195
4 cellTypescell_Stem 1.779897 0.138821
5 cellTypescell_TA 1.324436 0.145850
6 cellTypescell_TA.Early 1.944167 0.143707
7 cellTypescell_Tuft -0.654497 0.225239
8 condCond_1 0.178457 0.216167
9 cellTypescell_Enterocyte:condCond_1 1.026114 0.234573
10 cellTypescell_Enterocyte.Progenitor:condCond_1 -0.346556 0.250467
11 cellTypescell_Goblet:condCond_1 -0.033054 0.262500
12 cellTypescell_Stem:condCond_1 -0.659463 0.239814
13 cellTypescell_TA:condCond_1 -0.808348 0.257086
14 cellTypescell_TA.Early:condCond_1 -0.450709 0.241865
15 cellTypescell_Tuft:condCond_1 -0.527277 0.429530
statistic df p.value
0 25.533339 22.224333 0.000000e+00
1 8.689552 20.875604 2.240259e-08
2 10.821880 21.667775 3.348521e-10
3 3.846807 20.668018 9.586014e-04
4 12.821524 23.109362 5.442757e-12
5 9.080787 22.648182 5.234775e-09
6 13.528677 20.981438 7.842393e-12
7 -2.905783 21.966284 8.204102e-03
8 0.825554 21.753691 4.180192e-01
9 4.374399 21.402135 2.554530e-04
10 -1.383638 20.350230 1.814600e-01
11 -0.125920 22.708496 9.009037e-01
12 -2.749898 22.420741 1.156695e-02
13 -3.144273 22.698956 4.592254e-03
14 -1.863476 20.683645 7.665614e-02
15 -1.227567 20.908642 2.332512e-01
The simple Dirichlet-Multinomial model¶
Using the simple Dirichlet model is similar to using the scCODA model. Only HMC sampling is available for inference.
Significantly changing are all cell types where 0 is not included in the high density interval (HDI)
[10]:
K = data_salm.X.shape[1]
formula = "Condition"
cell_types = data_salm.var.index.to_list()
# Get count data
data_matrix = data_salm.X.astype("float32")
# Build covariate matrix from R-like formula
covariate_matrix = pt.dmatrix(formula, data_salm.obs)
covariate_names = covariate_matrix.design_info.column_names[1:]
covariate_matrix = covariate_matrix[:, 1:]
# Init model. Baseline index is always the last cell type
mod = om.SimpleModel(covariate_matrix=np.array(covariate_matrix), data_matrix=data_matrix,
cell_types=cell_types, covariate_names=covariate_names, formula=formula,
reference_cell_type=4)
# Run HMC sampling
result_simple = mod.sample_hmc()
result_simple.summary_extended()
2021-11-28 19:36:25.016681: W tensorflow/core/grappler/costs/op_level_cost_estimator.cc:689] Error in PredictCost() for the op: op: "Softmax" attr { key: "T" value { type: DT_DOUBLE } } inputs { dtype: DT_DOUBLE shape { unknown_rank: true } } device { type: "CPU" vendor: "GenuineIntel" model: "110" frequency: 2000 num_cores: 8 environment { key: "cpu_instruction_set" value: "AVX SSE, SSE2, SSE3, SSSE3, SSE4.1, SSE4.2" } environment { key: "eigen" value: "3.4.99" } l1_cache_size: 49152 l2_cache_size: 524288 l3_cache_size: 6291456 memory_size: 268435456 } outputs { dtype: DT_DOUBLE shape { unknown_rank: true } }
2021-11-28 19:36:25.038579: W tensorflow/core/grappler/costs/op_level_cost_estimator.cc:689] Error in PredictCost() for the op: op: "Softmax" attr { key: "T" value { type: DT_DOUBLE } } inputs { dtype: DT_DOUBLE shape { unknown_rank: true } } device { type: "CPU" vendor: "GenuineIntel" model: "110" frequency: 2000 num_cores: 8 environment { key: "cpu_instruction_set" value: "AVX SSE, SSE2, SSE3, SSSE3, SSE4.1, SSE4.2" } environment { key: "eigen" value: "3.4.99" } l1_cache_size: 49152 l2_cache_size: 524288 l3_cache_size: 6291456 memory_size: 268435456 } outputs { dtype: DT_DOUBLE shape { unknown_rank: true } }
MCMC sampling finished. (283.799 sec)
Acceptance rate: 69.1%
Compositional Analysis summary (extended):
Data: 6 samples, 8 cell types
Reference index: 4
Formula: Condition
MCMC Sampling: Sampled 20000 chain states (5000 burnin samples) in 283.799 sec. Acceptance rate: 69.1%
Intercepts:
Final Parameter HDI 3% HDI 97% SD \
Cell Type
Endocrine 1.043 0.307 1.726 0.387
Enterocyte 2.320 1.723 2.930 0.323
Enterocyte.Progenitor 2.483 1.834 3.075 0.334
Goblet 1.616 0.924 2.227 0.351
Stem 2.746 2.179 3.371 0.317
TA 2.161 1.467 2.807 0.355
TA.Early 2.849 2.233 3.440 0.324
Tuft 0.410 -0.382 1.213 0.431
Expected Sample
Cell Type
Endocrine 32.431823
Enterocyte 116.296171
Enterocyte.Progenitor 136.884860
Goblet 57.520427
Stem 178.063484
TA 99.200211
TA.Early 197.381842
Tuft 17.221182
Effects:
Final Parameter HDI 3% HDI 97% \
Covariate Cell Type
Condition[T.Salm] Endocrine 0.529575 -0.323 1.414
Enterocyte 1.608336 1.096 2.167
Enterocyte.Progenitor 0.329774 -0.243 0.936
Goblet 0.575193 -0.140 1.276
Stem NaN 0.000 0.000
TA -0.056400 -0.733 0.606
TA.Early 0.230121 -0.316 0.751
Tuft 0.133425 -0.972 1.171
SD Inclusion probability \
Covariate Cell Type
Condition[T.Salm] Endocrine 0.463 0.999200
Enterocyte 0.283 1.000000
Enterocyte.Progenitor 0.311 0.998333
Goblet 0.380 0.999333
Stem 0.000 0.000000
TA 0.356 0.998467
TA.Early 0.285 0.998200
Tuft 0.569 0.998800
Expected Sample log2-fold change
Covariate Cell Type
Condition[T.Salm] Endocrine 35.638644 0.136032
Enterocyte 375.850562 1.692356
Enterocyte.Progenitor 123.177940 -0.152219
Goblet 66.158172 0.201845
Stem NaN NaN
TA 60.670369 -0.709351
TA.Early 160.770249 -0.295989
Tuft 12.734065 -0.435491
Frequentist tests¶
Frequentist tests can all be used with the same scheme. Here, we use the ALR-transformed t-test as an example. The p.val attribute always contains the p-values of the test for all cell types (except the reference type if using ALR transforms).
[9]:
alr_t_model = om.ALRModel_ttest(data_salm, covariate_column="Condition")
alr_t_model.fit_model(reference_cell_type=4)
print(alr_t_model.p_val)
[0.061904043349529855, 0.016550016833625263, 0.01594225849767037, 0.4157960379583721, 1, 0.2886155729884671, 0.11388003371717678, 0.4785462482431266]