scCODA - Compositional analysis of single-cell data¶
This notebook serves as a tutorial for using the scCODA package (Büttner, Ostner et al., 2021) to analyze changes in cell composition data.
The package is intended to be used with cell composition from single-cell RNA-seq experiments, however there are no technical restrictions that prevent the use of data from other sources.
The data we use in the following example comes from Haber et al., 2017. It contains samples from the small intestinal epithelium of mice with different conditions.
This tutorial is designed to be executed on a standard computer (any operating system) in a Python environment with scCODA, Jupyter notebook and all their dependencies installed. Running the tutorial takes about 1.5 minutes on a 2020 Apple MacBook Pro (16GB RAM).
[13]:
# Setup
import importlib
import warnings
warnings.filterwarnings("ignore")
import pandas as pd
import pickle as pkl
import matplotlib.pyplot as plt
from sccoda.util import comp_ana as mod
from sccoda.util import cell_composition_data as dat
from sccoda.util import data_visualization as viz
import sccoda.datasets as scd
Data preparation¶
[14]:
# Load data
cell_counts = scd.haber()
print(cell_counts)
Mouse Endocrine Enterocyte Enterocyte.Progenitor Goblet Stem \
0 Control_1 36 59 136 36 239
1 Control_2 5 46 23 20 50
2 Control_3 45 98 188 124 250
3 Control_4 26 221 198 36 131
4 H.poly.Day10_1 42 71 203 147 271
5 H.poly.Day10_2 40 57 383 170 321
6 H.poly.Day3_1 52 75 347 66 323
7 H.poly.Day3_2 65 126 115 33 65
8 Salm_1 37 332 113 59 90
9 Salm_2 32 373 116 67 117
TA TA.Early Tuft
0 125 191 18
1 11 40 5
2 155 365 33
3 130 196 4
4 109 180 146
5 244 256 71
6 263 313 51
7 39 129 59
8 47 132 10
9 65 168 12
Mouse Endocrine Enterocyte Enterocyte.Progenitor Goblet Stem \
0 Control_1 36 59 136 36 239
1 Control_2 5 46 23 20 50
2 Control_3 45 98 188 124 250
3 Control_4 26 221 198 36 131
4 H.poly.Day10_1 42 71 203 147 271
5 H.poly.Day10_2 40 57 383 170 321
6 H.poly.Day3_1 52 75 347 66 323
7 H.poly.Day3_2 65 126 115 33 65
8 Salm_1 37 332 113 59 90
9 Salm_2 32 373 116 67 117
TA TA.Early Tuft
0 125 191 18
1 11 40 5
2 155 365 33
3 130 196 4
4 109 180 146
5 244 256 71
6 263 313 51
7 39 129 59
8 47 132 10
9 65 168 12
Looking at the data, we see that we have 4 control samples, and 3 conditions with 2 samples each. To use the models in scCODA, we first have to convert the data into an anndata object. This can be done easily with the sccoda.util.cell_composition_data module. The resulting object separates our data components: Cell counts are stored in data.X, covariates in data.obs.
[15]:
# Convert data to anndata object
data_all = dat.from_pandas(cell_counts, covariate_columns=["Mouse"])
# Extract condition from mouse name and add it as an extra column to the covariates
data_all.obs["Condition"] = data_all.obs["Mouse"].str.replace(r"_[0-9]", "", regex=True)
print(data_all)
AnnData object with n_obs × n_vars = 10 × 8
obs: 'Mouse', 'Condition'
AnnData object with n_obs × n_vars = 10 × 8
obs: 'Mouse', 'Condition'
For our first example, we want to look at how the Salmonella infection influences the cell composition. Therefore, we subset our data.
[16]:
# Select control and salmonella data
data_salm = data_all[data_all.obs["Condition"].isin(["Control", "Salm"])]
print(data_salm.obs)
Mouse Condition
0 Control_1 Control
1 Control_2 Control
2 Control_3 Control
3 Control_4 Control
8 Salm_1 Salm
9 Salm_2 Salm
Mouse Condition
0 Control_1 Control
1 Control_2 Control
2 Control_3 Control
3 Control_4 Control
8 Salm_1 Salm
9 Salm_2 Salm
Plotting the data, we can see that there is a large increase of Enterocytes in the infected sampes, while most other cell types slightly decrease. Since scRNA-seq experiments are limited in the number of cells per sample, the count data is compositional, which leads to negative correlations between the cell types. Thus, the slight decreases in many cell types might be fully caused by the increase in Enterocytes.
[17]:
viz.boxplots(data_salm, feature_name="Condition")
plt.show()
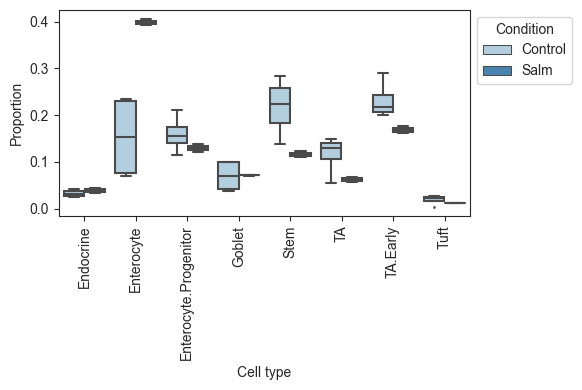
Note that the use of anndata in scCODA is different from the use in scRNA-seq pipelines, e.g. scanpy. To convert scanpy objects to a scCODA dataset, have a look at `dat.from_scanpy.
Model setup and inference¶
We can now create the model and run inference on it. Creating a sccoda.util.comp_ana.CompositionalAnalysis class object sets up the compositional model and prepares everxthing for parameter inference. It needs these informations:
The data object from above.
The
formulaparameter. It specifies how the covariates are used in the model. It can process R-style formulas via the patsy package, e.g.formula="Cov1 + Cov2 + Cov3". Here, we simply use the “Condition” covariate of our datasetThe
reference_cell_typeparameter is used to specify a cell type that is believed to be unchanged by the covariates informula. This is necessary, because compositional analysis must always be performed relative to a reference (See Büttner, Ostner et al., 2021 for a more thorough explanation). If no knowledge about such a cell type exists prior to the analysis, taking a cell type that has a nearly constant relative abundance over all samples is often a good choice. It is also possible to let scCODA find a suited reference cell type by usingreference_cell_type="automatic". Here, we take Goblet cells as the reference.
[18]:
model_salm = mod.CompositionalAnalysis(data_salm, formula="Condition", reference_cell_type="Goblet")
2022-06-27 10:33:10.230757: I tensorflow/core/platform/cpu_feature_guard.cc:151] This TensorFlow binary is optimized with oneAPI Deep Neural Network Library (oneDNN) to use the following CPU instructions in performance-critical operations: AVX2 AVX512F FMA
To enable them in other operations, rebuild TensorFlow with the appropriate compiler flags.
HMC sampling is then initiated by calling model.sample_hmc(), which produces a sccoda.util.result_classes.CAResult object.
[19]:
# Run MCMC
sim_results = model_salm.sample_hmc()
WARNING:tensorflow:@custom_gradient grad_fn has 'variables' in signature, but no ResourceVariables were used on the forward pass.
WARNING:tensorflow:@custom_gradient grad_fn has 'variables' in signature, but no ResourceVariables were used on the forward pass.
2022-06-27 10:33:13.334993: I tensorflow/compiler/xla/service/service.cc:171] XLA service 0x7f8509aca950 initialized for platform Host (this does not guarantee that XLA will be used). Devices:
2022-06-27 10:33:13.335016: I tensorflow/compiler/xla/service/service.cc:179] StreamExecutor device (0): Host, Default Version
0%| | 0/20000 [00:00<?, ?it/s]2022-06-27 10:33:13.435243: I tensorflow/compiler/mlir/tensorflow/utils/dump_mlir_util.cc:237] disabling MLIR crash reproducer, set env var `MLIR_CRASH_REPRODUCER_DIRECTORY` to enable.
2022-06-27 10:33:14.417583: I tensorflow/compiler/jit/xla_compilation_cache.cc:399] Compiled cluster using XLA! This line is logged at most once for the lifetime of the process.
100%|██████████| 20000/20000 [00:57<00:00, 348.77it/s]
MCMC sampling finished. (73.009 sec)
Acceptance rate: 55.6%
WARNING:tensorflow:@custom_gradient grad_fn has 'variables' in signature, but no ResourceVariables were used on the forward pass.
WARNING:tensorflow:@custom_gradient grad_fn has 'variables' in signature, but no ResourceVariables were used on the forward pass.
100%|██████████| 20000/20000 [00:51<00:00, 387.96it/s]
MCMC sampling finished. (69.511 sec)
Acceptance rate: 55.4%
Result interpretation¶
Calling summary() on the results object, we can see the most relevant information for further analysis:
[20]:
sim_results.summary()
Compositional Analysis summary:
Data: 6 samples, 8 cell types
Reference index: 3
Formula: Condition
Intercepts:
Final Parameter Expected Sample
Cell Type
Endocrine 1.109 34.615689
Enterocyte 2.325 116.781775
Enterocyte.Progenitor 2.508 140.233238
Goblet 1.739 64.994784
Stem 2.693 168.731218
TA 2.104 93.625897
TA.Early 2.856 198.602834
Tuft 0.422 17.414567
Effects:
Final Parameter Expected Sample \
Covariate Cell Type
Condition[T.Salm] Endocrine 0.000000 24.744607
Enterocyte 1.348671 321.590375
Enterocyte.Progenitor 0.000000 100.244037
Goblet 0.000000 46.460736
Stem 0.000000 120.615473
TA 0.000000 66.927342
TA.Early 0.000000 141.968837
Tuft 0.000000 12.448593
log2-fold change
Covariate Cell Type
Condition[T.Salm] Endocrine -0.484312
Enterocyte 1.461409
Enterocyte.Progenitor -0.484312
Goblet -0.484312
Stem -0.484312
TA -0.484312
TA.Early -0.484312
Tuft -0.484312
Compositional Analysis summary:
Data: 6 samples, 8 cell types
Reference index: 3
Formula: Condition
Intercepts:
Final Parameter Expected Sample
Cell Type
Endocrine 1.136 34.655473
Enterocyte 2.358 117.619596
Enterocyte.Progenitor 2.539 140.957109
Goblet 1.758 64.551003
Stem 2.717 168.419124
TA 2.129 93.546221
TA.Early 2.877 197.641672
Tuft 0.459 17.609802
Effects:
Final Parameter Expected Sample \
Covariate Cell Type
Condition[T.Salm] Endocrine 0.000000 24.773668
Enterocyte 1.343318 322.176411
Enterocyte.Progenitor 0.000000 100.764016
Goblet 0.000000 46.144663
Stem 0.000000 120.395398
TA 0.000000 66.872065
TA.Early 0.000000 141.285309
Tuft 0.000000 12.588470
log2-fold change
Covariate Cell Type
Condition[T.Salm] Endocrine -0.484276
Enterocyte 1.453722
Enterocyte.Progenitor -0.484276
Goblet -0.484276
Stem -0.484276
TA -0.484276
TA.Early -0.484276
Tuft -0.484276
Model properties
First, the summary shows an overview over the model properties: * Number of samples/cell types * The reference cell type. * The formula used
The model has two types of parameters that are relevant for analysis - intercepts and effects. These can be interpreted like in a standard regression model: Intercepts show how the cell types are distributed without any active covariates, effects show ho the covariates influence the cell types.
Intercepts
The first column of the intercept summary shows the parameters determined by the MCMC inference.
The “Expected sample” column gives some context to the numerical values. If we had a new sample (with no active covariates) with a total number of cells equal to the mean sampling depth of the dataset, then this distribution over the cell types would be most likely.
Effects
For the effect summary, the first column again shows the inferred parameters for all combinations of covariates and cell types. Most important is the distinctions between zero and non-zero entries A value of zero means that no statistically credible effect was detected. For a value other than zero, a credible change was detected. A positive sign indicates an increase, a negative sign a decrease in abundance.
Since the numerical values of the “Final Parameter” column are not straightforward to interpret, the “Expected sample” and “log2-fold change” columns give us an idea on the magnitude of the change. The expected sample is calculated for each covariate separately (covariate value = 1, all other covariates = 0), with the same method as for the intercepts. The log-fold change is then calculated between this expected sample and the expected sample with no active covariates from the intercept section. Since the data is compositional, cell types for which no credible change was detected, can still change in abundance as well, as soon as a credible effect is detected on another cell type due to the sum-to-one constraint. If there are no credible effects for a covariate, its expected sample will be identical to the intercept sample, therefore the log2-fold change is 0.
Interpretation
In the salmonella case, we see only a credible increase of Enterocytes, while all other cell types are unaffected by the disease. The log-fold change of Enterocytes between control and infected samples with the same total cell count lies at about 1.54.
We can also easily filter out all credible effects:
[21]:
print(sim_results.credible_effects())
Covariate Cell Type
Condition[T.Salm] Endocrine False
Enterocyte True
Enterocyte.Progenitor False
Goblet False
Stem False
TA False
TA.Early False
Tuft False
Name: Final Parameter, dtype: bool
Covariate Cell Type
Condition[T.Salm] Endocrine False
Enterocyte True
Enterocyte.Progenitor False
Goblet False
Stem False
TA False
TA.Early False
Tuft False
Name: Final Parameter, dtype: bool
Adjusting the False discovery rate¶
scCODA selects credible effects based on their inclusion probability. The cutoff between credible and non-credible effects depends on the desired false discovery rate (FDR). A smaller FDR value will produce more conservative results, but might miss some effects, while a larger FDR value selects more effects at the cost of a larger number of false discoveries.
The desired FDR level can be easily set after inference via sim_results.set_fdr(). Per default, the value is 0.05, but we recommend to increase it if no effects are found at a more conservative level.
In our example, setting a desired FDR of 0.4 reveals effects on Endocrine and Enterocyte cells.
[22]:
sim_results.set_fdr(est_fdr=0.4)
sim_results.summary()
Compositional Analysis summary:
Data: 6 samples, 8 cell types
Reference index: 3
Formula: Condition
Intercepts:
Final Parameter Expected Sample
Cell Type
Endocrine 1.109 34.615689
Enterocyte 2.325 116.781775
Enterocyte.Progenitor 2.508 140.233238
Goblet 1.739 64.994784
Stem 2.693 168.731218
TA 2.104 93.625897
TA.Early 2.856 198.602834
Tuft 0.422 17.414567
Effects:
Final Parameter Expected Sample \
Covariate Cell Type
Condition[T.Salm] Endocrine 0.288324 32.681545
Enterocyte 1.348671 318.351944
Enterocyte.Progenitor 0.000000 99.234574
Goblet 0.000000 45.992875
Stem 0.000000 119.400870
TA 0.000000 66.253380
TA.Early 0.000000 140.539204
Tuft 0.017884 12.545608
log2-fold change
Covariate Cell Type
Condition[T.Salm] Endocrine -0.082950
Enterocyte 1.446807
Enterocyte.Progenitor -0.498914
Goblet -0.498914
Stem -0.498914
TA -0.498914
TA.Early -0.498914
Tuft -0.473112
Compositional Analysis summary:
Data: 6 samples, 8 cell types
Reference index: 3
Formula: Condition
Intercepts:
Final Parameter Expected Sample
Cell Type
Endocrine 1.136 34.655473
Enterocyte 2.358 117.619596
Enterocyte.Progenitor 2.539 140.957109
Goblet 1.758 64.551003
Stem 2.717 168.419124
TA 2.129 93.546221
TA.Early 2.877 197.641672
Tuft 0.459 17.609802
Effects:
Final Parameter Expected Sample \
Covariate Cell Type
Condition[T.Salm] Endocrine 0.275744 32.882559
Enterocyte 1.343318 324.574906
Enterocyte.Progenitor 0.000000 101.514171
Goblet 0.000000 46.488195
Stem 0.000000 121.291701
TA -0.235592 53.229151
TA.Early 0.000000 142.337130
Tuft 0.000000 12.682187
log2-fold change
Covariate Cell Type
Condition[T.Salm] Endocrine -0.075761
Enterocyte 1.464423
Enterocyte.Progenitor -0.473575
Goblet -0.473575
Stem -0.473575
TA -0.813463
TA.Early -0.473575
Tuft -0.473575
Saving results¶
The compositional analysis results can be saved as a pickle object via results.save(<path_to_file>).
[23]:
# saving
path = "test"
sim_results.save(path)
# loading
with open(path, "rb") as f:
sim_results_2 = pkl.load(f)
sim_results_2.summary()
Compositional Analysis summary:
Data: 6 samples, 8 cell types
Reference index: 3
Formula: Condition
Intercepts:
Final Parameter Expected Sample
Cell Type
Endocrine 1.109 34.615689
Enterocyte 2.325 116.781775
Enterocyte.Progenitor 2.508 140.233238
Goblet 1.739 64.994784
Stem 2.693 168.731218
TA 2.104 93.625897
TA.Early 2.856 198.602834
Tuft 0.422 17.414567
Effects:
Final Parameter Expected Sample \
Covariate Cell Type
Condition[T.Salm] Endocrine 0.288324 32.681545
Enterocyte 1.348671 318.351944
Enterocyte.Progenitor 0.000000 99.234574
Goblet 0.000000 45.992875
Stem 0.000000 119.400870
TA 0.000000 66.253380
TA.Early 0.000000 140.539204
Tuft 0.017884 12.545608
log2-fold change
Covariate Cell Type
Condition[T.Salm] Endocrine -0.082950
Enterocyte 1.446807
Enterocyte.Progenitor -0.498914
Goblet -0.498914
Stem -0.498914
TA -0.498914
TA.Early -0.498914
Tuft -0.473112
Compositional Analysis summary:
Data: 6 samples, 8 cell types
Reference index: 3
Formula: Condition
Intercepts:
Final Parameter Expected Sample
Cell Type
Endocrine 1.136 34.655473
Enterocyte 2.358 117.619596
Enterocyte.Progenitor 2.539 140.957109
Goblet 1.758 64.551003
Stem 2.717 168.419124
TA 2.129 93.546221
TA.Early 2.877 197.641672
Tuft 0.459 17.609802
Effects:
Final Parameter Expected Sample \
Covariate Cell Type
Condition[T.Salm] Endocrine 0.275744 32.882559
Enterocyte 1.343318 324.574906
Enterocyte.Progenitor 0.000000 101.514171
Goblet 0.000000 46.488195
Stem 0.000000 121.291701
TA -0.235592 53.229151
TA.Early 0.000000 142.337130
Tuft 0.000000 12.682187
log2-fold change
Covariate Cell Type
Condition[T.Salm] Endocrine -0.075761
Enterocyte 1.464423
Enterocyte.Progenitor -0.473575
Goblet -0.473575
Stem -0.473575
TA -0.813463
TA.Early -0.473575
Tuft -0.473575
[23]: